![]()
MALDI-TOF
MS
Protein complexes isolated by the TAP affinity purification procedure
are boiled in SDS-PAGE loading buffer and separated by conventional SDS-PAGE.
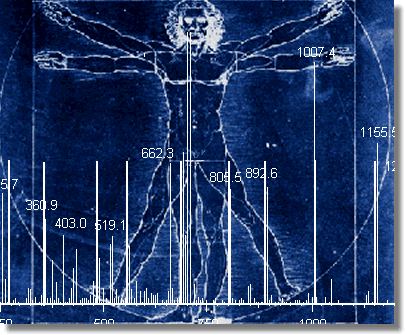
Gigital gel images are obtained and protein spots excised, in gel digested
with trypsin and the peptides extracted and analyzed by MALDI-TOF mass
spectrometry.
Tandem
MS
A powerful complimentary approach to analyze complex mixtures of low abundance
proteins, including the components of affinity purified protein complexes,
is centered on the direct analysis of trypsin-digested protein mixtures
using coupled tandem mass spectrometry (MS/MS) and sequence database searching.
Tandem mass spectrometry (MS/MS) provides a means for fragmenting a mass-selected ion and measuring the mass-to-charge ration (m/z) of the product ions that are produced during the fragmentation process. The MS/MS process used most often is based on collision-induced dissociation (CID), in which a mass-selected ion is transmitted to a high pressure region of the instrument where it undergoes low energy collisions with inert gas molecules. As a molecular ion collides, a portion of its kinetic energy is converted into excess internal energy rendering the ion unstable, and driving unimolecular fragmentation reactions prior to leaving the collision cell. Detailed structural information is generated as a result of fragmentation. The mass selectivity of many commercial MS systems permit the isolation of single precursor peptide ions from mixtures, thereby removing the contribution of any other peptide or contaminant from the sequence analysis step. The product ion spectra can subsequently be interpreted to deduce the amino acid sequence of a protein.
Tryptic peptides are particularly amenable to MS/MS analysis since mobile protons localize to the N-terminal amine and the side chains of the carboxy-terminal arginine or lysine residues at which proteolysis occurs. These protons cause peptides to fragment in a somewhat predictable manner following activation in a tandem MS, leading to production of two broad classes of fragment ions – the so-called amino-terminal b-type ions and carboxy-terminal y-type ions.
Very high resolution of protein mixtures can now be achieved by using multi-dimensional chromatographic approaches that separate peptides based on orthogonal strong cation exchange resin in the first dimension and by reverse phase fractionation in the second. The use of microcapillary scale chromatography online with tandem mass spectrometry allows the highest possible resolution of peptide mixtures (protein digests) and eliminates the need to separate proteins on polyacrylamide gels or to identify them using antibody-based techniques.
By allowing the routine identification of hundreds of proteins in a single run, this process enables a comprehensive characterization of the components of even the largest macromolecular assemblies, such as the ribosome, or the total protein complement of an organelle or an entire cell.