
ISRCTN72654148 |
Multiple Courses of Antenatal Corticosteroids for Preterm Birth Study
(MACS)
Study Protocol
December 2003
|
MULTIPLE COURSES OF ANTENATAL CORTICOSTEROIDS
FOR PRETERM BIRTH STUDY (MACS)
Data Coordinating Centre:
University of Toronto
Maternal, Infant and Reproductive Health Research Unit
at The Centre for Research in Women’s Health
7th Floor, 790 Bay Street
Toronto Ontario CANADA M5G 1N8
tel: 1-416-351-2530 fax: 1-416-351-2531
email: macs@sw.ca
TABLE OF CONTENTS
1. BACKGROUND
1.1 Introduction
1.2 Physiology
1.3 Pharmacology
1.4 Benefits and Risks of Single ACS
1.5 Benefits and Risks of Multiple ACS
1.6 Rationale for Proposed Research
2. PROTOCOL
2.1 Research Questions
2.2 Study Design
2.3 Selection Criteria for Participants
2.4 Selection Criteria for Centres
2.5 Schema
2.6 Randomisation
2.6.1 Prior to Randomisation
2.6.2 Randomisation
2.7 Manoeuvre
2.7.1 ACS Group
2.7.2 Placebo Group
2.7.3 Both Groups
2.8 Outcomes
2.8.1 Primary Outcome
2.8.2 Secondary Outcome
2.8.3 Additional Neonatal Outcomes
2.8.4 Additional 18-24 m Outcomes
2.8.5 Additional Maternal Outcomes
2.9 Methodologic Issues
2.9.1 Compliance
2.9.2 Contamination
2.9.3 Co-intervention
2.9.4 Masking
2.9.5 Non-randomised patients
2.9.6 Losses to Follow-up
2.10 Sample Size
2.11 Analysis
2.11.1 Interim Analysis
2.11.2 Final Analysis
2.11.3 Economic Analysis
2.12 Ethics
2.13 Feasibility
2.14 Time-line Diagram
2.15 Committee Structure
2.16 Data Management and Validation
2.17 Relevance
3. REFERENCES
1. BACKGROUND
1.1 Introduction
Preterm birth, delivery prior to 37 weeks gestational age, accounts for a major and disproportionate amount of infant morbidity and mortality.1 Today in Canada, approximately 7% of infants are born preterm.2 Despite advances in medical technology, the prevalence of preterm birth in Canada has actually increased. This appears to be secondary to an increase in multiple gestations, obstetric interventions, and ultrasound based estimates of gestational age.2 Fortunately, improvements have been made in regards to preterm neonatal morbidity and mortality, secondary to the advances in antenatal and neonatal care such as the use of antenatal corticosteroids (ACS) and surfactant.
Antenatal corticosteroids (ACS) were first used to enhance fetal lung maturation in 1972.3 Crowley (1990), summarised the results of 12 randomised controlled trials (RCTs) which demonstrated that ACS were highly effective in reducing rates of respiratory distress syndrome (RDS) and neonatal mortality.4 Four years later, the NIH held a consensus conference and summarised the multiple benefits of a single course of ACS in women at increased risk for preterm birth.5 Based on these results, it is widely accepted that a single course of ACS reduces morbidity and mortality in preterm infants and is indicated for most women at increased risk of preterm birth prior to 34 weeks gestation.
1.2 Physiology
Alveolar type II pneumocytes synthesise and secrete pulmonary surfactant, a complex substance comprised of lipids and proteins. Pulmonary surfactant maintains alveolar stability and normal lung function. Its deficiency in the new-born often leads to RDS. Corticosteroids are known to accelerate maturation of developmentally regulated proteins and to stimulate cytodifferentiation in numerous cells, including type II pneumocytes.6 In addition to increasing production of surfactant, corticosteroids increase lung compliance and maximal lung volume.7 Finally, corticosteroid treatment appears to reduce protein leak from the pulmonary vasculature into the airspace and appears to accelerate clearance of lung liquid prior to delivery.8 These effects represent precocious maturation and are essential in the transition to air breathing.8 It is unknown whether the beneficial effects of ACS on surfactant and other proteins are reversible. If the effects are reversible, it is unknown whether the levels of surfactant and these proteins return to pre-treatment levels or to new higher levels. At present, experimental evidence suggests that the induction of surfactant in fetal lung may be reversible.9,10 Transcription rates of surfactant proteins have been found to be significantly reduced, 4 hours following the removal of cortisol, in human fetal lung cultures.10
1.3 Pharmacology
The preferred ACS are dexamethasone and betamethasone. These corticosteroids readily cross the placenta in their biologically active forms, are weak in immunosuppressive activity, are devoid of mineralocorticoid activity and have a longer duration of action than cortisol.11 The bioavailability of non-synthetic corticosteroids to the fetus is reduced secondary to placental metabolism. The umbilical vein concentrations of betamethasone are approximately 25-30% of maternal venous concentrations.5 However, the corticosteroids do not remain in the fetal circulation for long. In one study, when the levels of betamethasone, administered prior to birth, were assayed in cord blood, the drug was undetectable forty hours following the injection.12
1.4 Benefits and Risks of a Single Course of ACS
The greatest benefit of a single course of ACS for fetuses at increased risk of preterm birth is a reduction in RDS. In the 2003 Cochrane meta-analysis by Crowley, the odds ratio for an effect of ACS on RDS was 0.53 (95% CI: 0.44, 0.63).13 ACS were also found to significantly reduce the risks of intraventricular haemorrhage (IVH), neonatal mortality, and the need for surfactant therapy. To date, follow up studies of infants enrolled in RCTs have not demonstrated any long-term adverse effects following a single course of ACS.14-19 One of these studies followed children to 12 years of age. There were no significant differences between the children that received a single course of ACS, as compared to those who did not, in terms of growth, or in terms of lung, neurologic or ophthalmologic function.17 There were also no differences in the children’s intellectual and motor development, school achievement and social-emotional functioning.16 When these children were followed to 20 years of age, no differences were found between the ACS and placebo groups as to medical and psychological variables.18 Indeed, the 2003 Cochrane review indicates a strong trend towards less risk of abnormal neurodevelopmental outcome among surviving children treated with ACS vs placebo (Odds ratio [95%CI]: 0.62 [0.36, 1.08]).13
The potential adverse maternal side effects of a single course of ACS include an increased risk of infection such as chorioamnionitis and endometritis. However, in Crowley’s meta-analysis, the frequency of maternal infection was similar between women who received and those who did not receive ACS, although, in one study, the rate of maternal infection was increased among women who received ACS who had preterm prolonged rupture of membranes (PPROM) > 24 hours.13,20
Although the effectiveness of one course of ACS (greater benefit than risk for those treated) has been proven, there is uncertainty as to how long the treatment continues to be effective if the woman remains undelivered 7 or more days following the initial dose. When the Liggins trial was reported in 1972, they reported the risk of RDS among liveborn infants after unplanned premature labour, born at <2 days, 2-<7 days and ³7 days. They did not report the effects of ACS for those born at 7-14 days or at other intervals of time following the initial dose. They found that the risk of RDS was significantly reduced following treatment with ACS (vs control) among fetuses born between 2 days and less than 7 days following trial entry (3.6% vs 33.3%, p=0.03), but the effect on RDS was not statistically significant following treatment with ACS (vs control) if fetuses were born 7 or more days following trial entry (2.2% vs 9.4%, p>0.05).3 The Collaborative Group on Antenatal Steroid Therapy also reported their results for singleton infants in this way. They found the risk of RDS to be reduced with ACS (vs placebo), if delivery occurred 24 hours – 7 days following trial entry (9.3% vs 20.1%), and if delivery occurred >7 days following trial entry (6.0% vs 10.5%).21 The question has arisen, therefore, as to whether the effectiveness of the ACS is lost or reduced if the woman remains undelivered 7 days after treatment.
Because of this, and because the risk of RDS and other complications of prematurity are high for fetuses born very preterm, some clinicians have suggested that weekly courses of ACS should be given to women who are at increased risk of preterm birth and remain undelivered more than 7 days following the initial dose.22 In the 1980’s and 1990’s, this approach became routine in some medical centres, despite the fact that multiple courses of ACS had not been evaluated in well-designed RCTs and the benefit-to-risk ratio was therefore, not known. In a survey in 1997, in the UK, 98% of respondents indicated they prescribed repeated courses of antenatal corticosteroids for women who remained at increased risk of preterm birth.23 In a similar survey of Australian obstetricians, 50-85% of obstetricians indicated they prescribed multiple courses of ACS.24 Despite the theoretical possibility that the effectiveness of a single course of ACS may be lost if the woman remains undelivered more than 7 days following the initial dose, the 2003 Cochrane meta-analysis actually shows a reduction in RDS for these fetuses (N=265) (odds ratio: 0.41), albeit of only borderline statistical significance (95% CI: 0.18, 0.98), and this odds ratio is actually of a similar magnitude to that for fetuses born between 24 hours and 7 days following the initial dose (N=728) (odds ratio: 0.38, 95% CI: 0.25, 0.57).13 However, because fewer mothers/fetuses delivering more than 7 days following the initial dose were included in this meta-analysis, and because the overall risk of RDS for those infants born later was lower, the 95% confidence interval about the odds ratio is wide. Thus it is possible that the beneficial effects of a single course of ACS continue beyond 7 days.
1.5 Benefits and Risks of Multiple Courses of ACS
There are insufficient data available from appropriately powered well-designed RCTs as to the benefits and risks of multiple courses of ACS. Data from one RCT by Guinn et al, of 502 women randomised to single vs. weekly courses of ACS, showed no difference in the primary outcome (a composite of RDS, bronchopulmonary dysplasia [BPD], neonatal sepsis, necrotizing enterocolitis [NEC] or neonatal death) between the infants who received single vs. weekly courses of ACS, although the trend was toward benefit (28.0% vs 22.5%, p=0.16).25 This trial was stopped early before reaching its planned sample size and thus lacks power for finding clinically important reductions in adverse perinatal outcomes. Despite its early closure, Guinn et al reported a statistically significant reduction in risk of severe RDS in the weekly ACS group and a planned subgroup analysis showed a significant decrease in composite morbidity among neonates in the ACS group, delivered prior to 28 weeks’ gestation.25 Two other small RCTs of weekly ACS vs placebo have been published, but the sample sizes were too small to add to our information as to benefits and risks of weekly courses of ACS.26,27 The data from these three RCTs have been included in a Cochrane Review, which concludes that additional RCTs are needed.28
A systematic review of retrospective and other non-randomised studies in humans has suggested that multiple courses of ACS may decrease the risk of RDS and patent ductus arteriosus without evidence of adverse neonatal effects, compared to one course.29 One non-randomised study followed children to 7 years of age and did not find multiple courses of ACS to be associated with adverse effects.30 However, because these studies were not randomised, confounding factors make the findings difficult to interpret. Given the known benefits of a single course of ACS (i.e. decreased risk of RDS, IVH and neonatal mortality), it follows that multiple courses of ACS could continue to benefit an infant if the pregnancy is at increased risk of preterm birth and remains undelivered more than 7 days following the initial dose. Animal data are consistent with this hypothesis, as studies in animals have found progressive improvement in postnatal lung function following multiple (vs single) courses of ACS.31
However, adverse effects of multiple courses of ACS have been reported in animals.31
Multiple courses of ACS have been associated with a decrease in birth weight (sheep, rabbits, mice), and a delay in the maturation or development of the fetal nervous system (sheep, monkeys).31 In addition, multiple courses of ACS have been associated with permanent changes in the expression of glucocorticoid receptors in the hippocampus and limbic system, suggestive of a decrease in central glucocorticoid feedback (rats and guinea pigs).32,33
Also, there is some evidence that postnatal corticosteroids (PCS) given directly to infants after birth, may increase the risk of long-term neurologic impairment.34
If any of these adverse effects are true for ACS, when given to human fetuses, it is possible that they are dose dependent, that is, the more corticosteroid that is given, the greater may be the risk. Thus if there are truly benefits and risks to multiple courses of ACS in humans, less rather than more frequent administration may optimise the benefit-to-risk ratio.
1.6 Rationale for Proposed Research
A favourable benefit-to-risk ratio is well established for a single course of ACS.13 However, insufficient evidence exists in regards to the effectiveness of multiple courses of ACS. There are currently inadequate data from RCTs to know the true benefits and risks of multiple versus single courses of ACS. Despite this fact, many centres routinely prescribe multiple courses of ACS to women who are at increased risk of preterm birth and remain undelivered for greater than 7 days following the initial dose. For many centres the repeated courses are given at weekly intervals. For others, particularly those who are concerned about the risk of adverse effects, a more conservative approach is followed and repeated courses are given every 14 days or at different points in gestation, such as at 26 and 28 weeks.
As of December 2003, two other multicentre RCTs of weekly courses of ACS have been completed (results not yet published); the UK trial (N=154) and the NICHD trial (N=492). Another multicentre trial of weekly ACS, with a sample size of 980, is underway in Australia/New Zealand. In each RCT, the intervention includes repeating the courses of ACS at 7-day intervals. As we wish to minimise the potential for harm from the administration of this quantity of ACS, we are proposing to study the effect of ACS at less frequent intervals, that is, every 14 days. In our study, we will only include women at highest risk for the adverse outcomes of prematurity, those <33 weeks gestation. We will follow children to 18-24 months of age and a proposal for a further follow-up to 5 years of age is planned. We anticipate that this study will be complementary to the others and will help answer the question as to the proper place, if any, of multiple courses of ACS for women who remain undelivered and continue to be at increased risk of preterm birth after the initial treatment.
2. PROTOCOL
2.1 Research Questions
Primary Research Question:
For women at 250/7-326/7 weeks gestation, who remain at increased risk of preterm birth†, 14-21 days following a single course of ACS, do multiple courses of ACS, every 14 days, until 336/7 weeks, decrease (or increase) the risk of perinatal or neonatal mortality or significant neonatal morbidity‡, compared to placebo? (†see inclusion criteria (2.3) for definition, ‡see primary outcome (2.8.1) for definition)
Secondary Research Question:
For women at 250/7-326/7 weeks gestation, who remain at increased risk of preterm birth†, 14-21days following a single course of ACS, do multiple courses of ACS, every 14 days, until 336/7 weeks, decrease (or increase) the risk of neurologic impairment of children at 18-24 months of age (corrected for gestational age at birth), compared to placebo? (See secondary outcome (2.8.2) for definition.)
Additional Research Questions:
For women at 250/7-326/7 weeks, who remain at increased risk of preterm birth†, 14-21 days following a single course of ACS:
- Are multiple courses of ACS, every 14 days until 336/7 weeks, associated with higher or lower a) birth weight, b) birth length, c) birth head circumference, d) length of stay in neonatal intensive care and, e) number of days of intubation assisted ventilation, f) patent ductus arteriosus (PDA) requiring pharmacological treatment or surgery, compared to placebo?
- Do multiple courses of ACS, every 14 days until 336/7 weeks, increase or decrease the risk of a) neonatal infection, and b) retinopathy of prematurity (ROP), compared to placebo?
- Do multiple courses of ACS, every 14 days until 336/7 weeks, increase or decrease the risk of: a) clinical chorioamnionitis and, b) antepartum or postpartum maternal infection (pneumonia, endometritis, wound infection, sepsis, pyelonephritis), compared to placebo?
- Do multiple courses of ACS, every 14 days until 336/7 weeks, increase or decrease the risk of postpartum depression (to be determined by the Edinburgh Postnatal Depression Scale), to be administered by a questionnaire at 3 months postpartum, compared to placebo?
- Do multiple courses of ACS, every 14 days until 336/7 weeks, increase maternal side effects (headache, moonface, acne, excess hair growth, swelling, striae, sleeping difficulty, muscle weakness, increased appetite, bruising, memory problems, mood swings), compared to placebo?
- Are multiple courses of ACS, every 14 days until 336/7 weeks, associated with higher or lower height, weight, or head circumference of the children at 18-24 months of age, compared to placebo?
2.2 Study Design
The study is a multicentre, double masked, RCT, with prognostic stratification for gestational age (250/7-276/7 weeks; 280/7-326/7 weeks) and centre. Randomisation will be centrally controlled using a telephone computerised randomisation service. Eligible and consenting women will be randomised within centre and by gestational age groups to receive additional courses of ACS or placebo, using block sizes of 4. A double masked RCT is the best study design to answer this question as it avoids selection and outcome assessment bias and will minimise contamination.35
2.3 Selection Criteria for Participants
The target population is women at increased risk of preterm birth†, at 250/7-326/7 weeks gestation, who have received one course of ACS, 14-21 days ago. Women will not be eligible until they reach 250/7 weeks gestation, as we do not expect an initial course of ACS to be given prior to 230/7 weeks gestation. Women who receive an initial course of antenatal corticosteroids prior to 230/7 weeks gestational age will be considered ineligible.
Inclusion Criteria:
- Women who have previously received one completed course of ACS§, 14-21 days ago and continue to be at increased risk of preterm birth†
- Gestational age greater than or equal to 250/7 weeks of gestation and less than or equal to 326/7 completed weeks of gestation [gestational age will be determined by the clinician using menstrual history and early ultrasound if available]
- All fetuses must be alive at randomisation. However, in multiple gestation pregnancies, if any fetus is thought to have died prior to 13 weeks, that fetus will not be considered part of the pregnancy for the purposes of this study.
§To be
eligible for trial entry women will have received one completed course of ACS (eg: 2 doses of intramuscular betamethasone, 12 mg/dose, given at 12 or 24 hour intervals; 4 doses of intramuscular betamethasone 6mg/dose given at 12 hour intervals; 4 doses of intramuscular dexamethasone, 5-6 mg/dose, given at 12 hour intervals; 2 doses dexamethasone, 12 mg/dose, given at 12 hours intervals).
†As a guideline, women at increased risk of preterm birth might have one or more of the following: uterine contractions within the previous week, a shortened cervical length or cervical dilation, PPROM, antepartum bleeding secondary to placental separation or placenta previa, a history of preterm delivery, multiple pregnancy, maternal hypertension or other medical condition requiring preterm delivery, or intrauterine growth restriction or other fetal condition requiring preterm delivery. If the clinical condition is of sufficient concern that the woman would be given one course of ACS, the clinical condition is to be considered at increased risk of preterm birth.
Exclusion Criteria:
- Women requiring chronic doses of corticosteroids secondary to medical conditions (e.g. systemic lupus erythematosus, congenital adrenal hyperplasia, severe asthma)
- Women with a contraindication to corticosteroids
- Women with clinical evidence of chorioamnionitis (temperature ³ 38°C)
- Known lethal congenital anomaly (e.g. anencephaly) in any fetus
- First course of ACS given prior to 23 weeks
- Previous participation in MACS.
Note that PPROM is not an exclusion to participation, because there is no evidence that the benefits of multiple courses of ACS would not apply to women with PPROM, and because there is no substantive evidence for an increased risk of maternal or fetal infection following ACS for women with PPROM.13
2.4 Selection Criteria for Centres
Hospital centres will be invited to participate in the study if they are confident that they can achieve a follow-up rate of >80% for assessing neurodevelopmental outcome at 18-24 months of age.
2.5 Schema
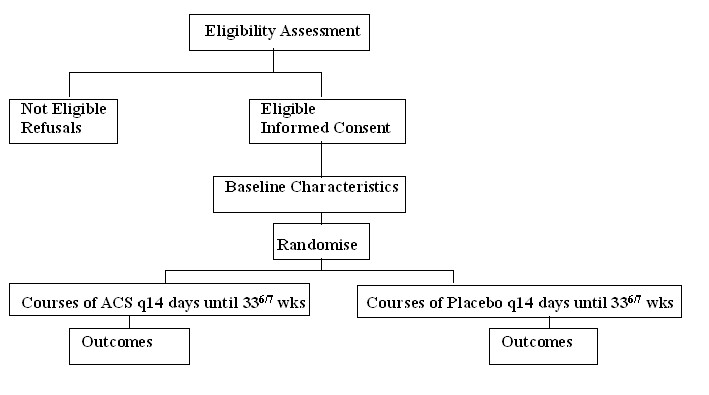 2.6 Randomisation
2.6 Randomisation
2.6.1 Prior to Randomisation
Within the 24 hours preceding enrolment in the study, women will have a non-stress test or biophysical profile to rule out fetal compromise, and their temperature taken to rule out clinical evidence of chorioamnionitis. An obstetrical ultrasound will be performed within the 2-3 weeks prior to randomisation to determine the number of fetuses, the estimated weight of the fetus(es), the presence of lethal or other congenital anomalies, the presence of placental anomalies, and the adequacy of the amniotic fluid. When women are identified as being at increased risk of preterm birth, and are given their initial course of ACS, they will be informed about MACS in the event they remain undelivered 14 days later and continue to be at increased risk of preterm birth. A participant information sheet, outlining the possible benefits and risks of additional courses of ACS and the details of MACS, will be provided (see Appendix 1). Eligible women will be invited to participate in the study. Women who agree to participate will sign a consent form (see Appendix 1), and baseline information will be collected prior to randomisation.
2.6.2 Randomisation
Women will be randomised using the centrally controlled computerised telephone randomisation service at the University of Toronto Maternal, Infant & Reproductive Health Research Unit (MIRU). Using the touch pad of the telephone, eligibility and baseline information will be recorded during the telephone call. Following this, a treatment number will be issued. The treatment number will correspond to a study box at the centre, and will contain vials of betamethasone or corresponding placebo. The vials supplied will be of similar appearance.
2.7 Manoeuvre
2.7.1 ACS Group
Women allocated to a treatment number corresponding to ACS will receive a course of ACS, which will consist of two doses, 12 mg per dose, of betamethasone given intramuscularly 24 hours apart. The betamethasone formulation is a combination of betamethasone phosphate and acetate, and will be supplied by Schering-Plough Corporation, Madison, New Jersey, USA. Following this, if the woman remains at increased risk of preterm birth, she will continue to receive multiple courses of ACS every 14 days until 336/7 weeks gestation. However, if a woman has PPROM one should consider stopping the study medication earlier, such as 326/7 weeks. All patients and care-givers will remain masked to the actual treatment given.
2.7.2 Placebo Group
Women allocated to a study number corresponding to placebo will receive a course of placebo, which will consist of two doses of a placebo of similar appearance, given intramuscularly 24 hours apart. The placebo will consist of a dilute concentration of aluminum monostearate. This substance is commonly used as a filler in many pharmaceutical preparations and is considered to be inert. The placebo will be supplied by Eminent Services Corporation, Gaithersburg, Maryland, USA. Following this, if the woman remains at increased risk of preterm birth she will receive multiple courses of placebo every 14 days until 336/7 weeks gestation. However, if a woman has PPROM one should consider stopping the study medication earlier, such as 326/7 weeks. All patients and care-givers will remain masked to the actual treatment given.
2.7.3 Both Groups
Centres will be encouraged to treat women with antibiotics if they develop clinical signs of chorioamnionitis, or if they are in preterm labour and either they are known to be colonised with group B streptococcus (GBS) or their GBS status is unknown. As there is good evidence now to suggest an association between the use of postnatal corticosteroids and an increased risk of neurodevelopmental impairment, centres are encouraged
not to use postnatal corticosteroids.34 The quantity and duration of postnatal corticosteroid use will be documented.
If infants are born less than 1500 grams, centres should arrange at least two cranial ultrasounds prior to hospital discharge to look for evidence of IVH and cystic periventricular leukomalacia (PVL). We recommend that one cranial ultrasound is performed in the first week and a second between the second and fourth weeks of life. Centres are also encouraged to perform ophthalmologic exams to look for retinopathy of prematurity (ROP) for infants born less than 1500 grams.
All other aspects of medical care for mothers and their infants will be determined by the treating physician, according to local hospital policies. Data will be collected to describe maternal/fetal and neonatal treatments, such as the use of tocolytics prior to delivery, and the use of neonatal treatments, such as surfactant, antibiotics, corticosteroids, and indomethacin.
All mothers will be asked to complete a structured questionnaire 3 months following birth to determine the occurrence of postpartum depression and maternal side effects.
All babies will be followed until 18-24 months of age (corrected for gestational age at birth), at which time they will undergo neurodevelopmental and behavioural assessments. The assessments will include an assessment of motor function (Gross Motor Function Classification System [Palisano et al]),36 and a standardised neurological examination to rule out cerebral palsy (CP),36 documentation of previous hospitalizations, height, weight, head circumference, a clinical impression of developmental level and an assessment using the revised Bayley Scales of Infant Development (BSID-II) to determine the level of mental and physical functioning (Mental Developmental Index [MDI] and Physical Developmental Index [PDI]) and to assess behaviour (BRS). The BSID-II are well standardised, and have demonstrated reliability and validity.37 If the BSID-II are not available locally, then the results of other standardised tests may be accepted. Personnel for the neurodevelopmental assessments will include neonatologists, general paediatricians, developmental paediatricians, or trained nurses, who are experienced in performing neurodevelopmental examinations for follow-up programs, development assessment centres &/or treatment centres for disabilities. The BSID-II will be administered by an individual who has been specifically trained in the administration of the test.
2.8 Outcomes
2.8.1 Primary Outcome: Perinatal or neonatal mortality or significant neonatal morbidity
Perinatal or neonatal mortality is defined as stillbirth, or neonatal death during the first 28 days of life or prior to hospital discharge, whichever is later.
Significant neonatal morbidity is defined as one or more of the following:
- Respiratory Distress Syndrome (RDS): defined as requiring assisted ventilation via endotracheal tube and supplemental oxygen both within the first 24 hours of life and for a duration of greater than or equal to 24 hours, and either an x-ray compatible with RDS or surfactant given between the first 2 and 24 hours of life.
- Bronchopulmonary dysplasia (BPD) (defined as requiring oxygen at a postnatal gestational age of 36 completed weeks and X-ray compatible with BPD)
- IVH grade III or IV, diagnosed by cranial ultrasound, using categorisation of Papile et al38, or at autopsy
- Cystic PVL (defined as periventricular cystic changes in the white matter, excluding sub-ependymal and choroid plexus cysts) diagnosed by cranial ultrasound or at autopsy
- NEC (defined as either perforation of intestine, pneumatosis intestinalis or air in the portal vein) diagnosed by X-ray, surgery, or at autopsy.39
2.8.2 Secondary Outcome: Death or neurologic impairment at 18 to 24 months of age (corrected for gestational age at birth)
Death or neurologic impairment at 18 to 24 months of age (adjusted for gestational age at birth) is defined as one or more of the following:
- Death
- Cerebral palsy (CP)36
- BSID-II (MDI) <7037 (that is: more than two standard deviations below the norm, or the local equivalent on other standardised tests)
>2.8.3 Additional Neonatal Outcomes
- Birth weight, birth length and birth head circumference
- Neonatal infection (clinical signs of infection and one or more of the following: a positive culture of blood, cerebrospinal fluid [CSF], or lung tissue at autopsy; a positive Gram stain of CSF; a chest X-ray compatible with pneumonia; or a histological diagnosis of pneumonia at autopsy)
- Retinopathy of prematurity (ROP), diagnosed in one or both eyes
- Length of stay in neonatal intensive care and duration of use of ventilation with intubation
- PDA (patent ductus arteriosus) requiring pharmacological treatment or surgery
2.8.4 Additional 18 to 24 month Outcomes (corrected for gestational age at birth)
- Height
- Weight
- Head circumference
2.8.5 Additional Maternal Outcomes
- Clinical chorioamnionitis (defined as maternal temperature ³ 38°C prior to delivery and one or more of the following: maternal tachycardia ³120 bpm, white blood cell count ³20,000/mm3, fetal tachycardia >160 bpm, uterine tenderness, or foul smelling amniotic fluid)
- Maternal infection (defined as one or more of the following: endometritis [postpartum maternal temperature ³38°C and tender fundus without other source of infection], pneumonia [maternal temperature ³38°C and signs of pneumonia on X-ray], wound infection or wound breakdown, pyelonephritis [maternal temperature ³38°C, positive urine culture and costal vertebral angle tenderness], or sepsis [maternal temperature ³38°C and a positive maternal blood culture]
- Postpartum depression defined as a score >12 on the Edinburgh Postnatal Depression Scale at 3 months postpartum40
- Maternal side effects (headache, moonface, acne, excess hair growth, swelling, striae, sleeping difficulty, muscle weakness, increased appetite, bruising, memory problems, mood swings).
2.9 Methodologic Issues
2.9.1 Compliance
Women in the study should not be given antenatal corticosteroids to promote fetal lung maturity apart from the study drugs. Women will be given a booklet and will be instructed to carry it with them. The booklet will contain the details of the study protocol, their study number, the dates and times of courses of study drug received and when future courses are required. If a participant is discharged from hospital, the woman will be given her remaining study drugs to take with her to give to her referring physician. Therefore, if women are discharged from a referral hospital, compliance with the protocol can be assured. Referring physicians in each centre will be informed about the study, generally, and specifically before any of their patients are randomised. Study packs will be returned to the participating centres when the woman reaches 340/7 weeks gestation to confirm the use of study medication. The study coordinator in each centre will maintain ongoing contact with each participant and will track participants weekly, to assure that they receive the study drug, or that there is a valid reason why the study drug is not given. Data on compliance regarding treatment will be sent regularly to the Data Centre where centre compliance will be tracked.
2.9.2 Contamination
Contamination is unlikely to be a problem because physicians and participants will be masked to the group allocation. To minimise study patients receiving ACS outside the study protocol, women in the study will be given a booklet to carry with them, which will identify their involvement in MACS. Copies of this information will be sent to the referring physician and will be available to the physician treating the patient.
2.9.3 Co-intervention
Information regarding co-interventions will be collected on the data forms. Co-interventions include maternal antibiotics, tocolytic agents, and neonatal treatments such as surfactant, antibiotics, corticosteroids, and indomethacin. Co-interventions will likely be similar between groups as the physicians and patients will not be aware of the group allocation, unless the co-interventions are a result of the effect of the multiple courses of ACS on the mother or infant.
2.9.4 Masking
To the extent possible, physicians and patients will be masked to group of allocation until the study is completed. However, we recognise that ACS may cause maternal hyperglycaemia, which may unmask caregivers, particularly if a woman has diabetes. Centres will be counselled to assume that all women are receiving ACS if they are concerned about side effects and to monitor women in the study accordingly. If adverse effects occur such that continuation of ACS is contraindicated, the study drug will be discontinued, regardless of the group the woman has been allocated to. Thus masking in this situation, will be maintained. If a complication develops, in which the subsequent care of the patient is dependent on knowing the treatment assignment, arrangements will be made to allow for unmasking. The study research coordinator (or designate) in the Data Centre will be available by pager, 24 hours/day, 7 days/week, to provide the details regarding an individual’s allocation group, should this be necessary.
2.9.5 Non-randomised patients
There is no reason to believe that the effect of multiple courses of ACS will differ in randomised versus non-randomised patients and we do not believe the extra cost of obtaining baseline and outcome information on non-randomised patients, in this study, can be justified.
2.9.6 Losses to Follow-up
Following randomisation, there are two occasions when study participants may be lost to follow-up. The first time is after the high risk condition improves and the woman is referred back to her local physician for continuing care. Should this occur, centres will be asked to have a system in place to continue to track participants through to delivery to ensure that the study drugs are administered, if appropriate, and that the data are collected at the relevant times. Study participants may also be lost to follow-up at the time of the 18 to 24 month assessment. As centres will only be eligible to participate if they have experience with follow-up, such that they are confident they can track at least 80% of randomised patients, we do not anticipate that loss to follow-up will be a problem. We will ask centres to record various contact details (e.g. mother’s mother’s contact details, a friend’s contact details, referring physician’s contact details etc.) to facilitate the tracking of participants, should patients move. Centres will be provided with regular quarterly reminders, to send to participants to ask them to contact the participating centre, should they move or change their telephone number or other contact information.
2.10 Sample Size
The sample size has been calculated to be 1900 women (950/group). A sample size of 1900 women will have 80% probability of achieving statistical significance if multiple courses reduces the risk of RDS from 12% to 8% (2-tailed a error of 0.05).
In calculating the sample size we have assumed the following:
- The rate of RDS would be a reasonable surrogate measure for estimating the rate of perinatal or neonatal mortality or serious neonatal morbidity in this study.
- The rate of RDS is approximately 12% for infants in the placebo group. This is based on the information from the Ballard trial,41 in which women at risk of preterm birth were enrolled at <30 weeks gestation to receive thyrotropin hormone (TRH) plus ACS versus placebo plus ACS (1 course). For those infants in the placebo group, who were born >10 days following entry to the study, the rate of RDS was 19%. As we are enrolling women at <33 weeks gestation who will be at 14 days following the initial treatment of ACS, we have estimated the rate of RDS to be 12% in the placebo group.
- The rate of cerebral palsy (CP) in the Low Birth Weight (<1250g) Follow-up Clinics at the Hospital for Sick Children and at McMaster University is approximately 10%. Assuming that half of the children in this cohort will be delivered near term, the rate of CP would approximate 5%. Assuming that CP is the most frequently occurring neurologic deficit in these children, a sample size of 1900 will have greater than 90% power to detect an increase in the incidence of neurologic impairment from 5% to 9% (2-tailed a error of 0.05), if such an effect exists.
2.11 Analyses
2.11.1 Interim Analysis
After complete data have been received on the first 800 patients enrolled, an interim analysis will be undertaken. This will include an analysis of the important baseline variables and an analysis of the primary outcome. The data will be presented for each group using an intention to treat approach, masked to knowledge of actual group. The interim analysis will be presented to an independent Data and Safety Monitoring Board for their advice as to whether the study should be stopped early. The decision to stop the trial will be based on finding a higher rate of the primary outcome in the ACS group versus the placebo group (P<0.002, 1 tailed test). A one-sided test will be performed as we intend to stop the study early only if we can conclude that ACS is detrimental. Otherwise the trial will continue until the sample size has been achieved. The study will not be stopped if multiple courses of ACS are found to be beneficial, as the corresponding information regarding the 18 to 24 month secondary outcome will not yet have been collected.
2.11.2 Final Analysis
The final analysis of the outcome data will be based on an “intention to treat” approach, which will include all patients as randomised. Descriptive statistics will be calculated to check for any major dissimilarities in the study groups with regard to patient demographics, prognostic and other baseline information.
Logistic regression will be used to calculate the adjusted odds ratios and 95% confidence intervals for the comparison of the two study groups with respect to the primary outcome, perinatal or neonatal mortality, or significant neonatal morbidity, and the secondary outcome, death or neurologic impairment at 18 to 24 months of age, controlling for possible prognostic factors (gestational age at enrolment, singleton vs. multiple pregnancy, PROM, etc.). Random effects models will be used to determine if treatment effects vary between centres. The level of statistical significance for the analysis of the primary and secondary outcomes will be P<0.05 using a 2-tailed test. Outcome rates will be reported as the proportion of infants affected. However, because outcomes between infants in a multiple pregnancy are correlated, the statistical analysis will be based on the pregnancy as the unit of analysis. Therefore, if any infant in a multiple pregnancy has the outcome, then the pregnancy will be deemed to have the outcome. In addition, rate differences will be used to determine the number needed to treat.
Logistic regression will also be used to calculate the adjusted odds ratios and 95% confidence intervals for the comparison of the two study groups with respect to the other neonatal and maternal binary outcomes. Analysis of covariance will be used to compare the group’s continuous outcome measures, controlling for baseline demographic and prognostic characteristics. The level of statistical significance for these secondary analyses will be P<0.01. Continuous outcome measures that are skewed will undergo the appropriate transformation (most likely log or square root) prior to analysis.
2.11.3 Economic Analysis
The clinical hypothesis is that multiple course of ACS are more effective and less expensive than placebo. Therefore, the economic hypothesis is that multiple courses of ACS are less expensive than placebo. The economic analysis will involve determining the resources consumed and unit prices for these resources. Resources consumed in either intervention in the trial will be collected in the clinical case report forms.
The estimation of unit prices in this trial will be obtained from a sample of six participating Canadian hospitals. Hospitals will be chosen by the following criteria: a) hospitals with available established costing data, b) hospitals representative in various Canadian geographic locations, c) small and large centres. For each hospital we will develop a costing model based on the available data within the hospital. The estimates of unit prices will be combined with the resource use information for all trial participants.
2.12 Ethics
When women are identified as being at increased risk of preterm birth, and are given their initial course of ACS, they will be informed about MACS in the event they remain undelivered at 14-21 days and continue to be at increased risk of preterm birth. A participant information sheet, outlining the possible benefits and risks of additional courses of ACS and the details of MACS, will be provided. Eligible women will be invited to participate. Women who agree to participate will sign consent prior to randomisation. It will be reinforced that participants can withdraw from the trial at any time and that a woman’s care will not be jeopardised if she should choose not to participate. The “Information Sheet” and “Consent Form” are included in Appendix 1.
2.13 Feasibility
We estimate that approximately 0.8% of all births at participating centres will be at increased risk of preterm birth at <310/7 weeks gestation and will consent to participate. This estimate is based on the actual recruitment rate into the Knight TRH Trial at National Women’s Hospital, Auckland, New Zealand42 in which approximately 1.1% of all births were at increased risk of preterm birth at <33 weeks gestation and consented to that study. We estimate that 40% of these women will remain undelivered 14 days later and will therefore be eligible for this trial. This estimate is based on the Ballard trial41 in which 50% of women enrolled at <30 weeks gestation remained undelivered 10 days following enrolment. Thus, we expect to recruit (0.8% x 0.4 = 0.32%) 0.32% of women in participating centres to the study. If, during the first 6 months of enrolment, the recruitment rate averages 0.25%, and then 0.32% after that, we will need centres with a total of approximately 190,000 deliveries to be able to recruit 1900 women in 3-4 years.
2.14 Time-line Diagram
Apr 00 – Dec 00
* Develop a manual of operations for the centres
* Develop a manual of operations for The Data Centre
* Develop and present data forms and distribute to centres
* Organise the trial in participating centres
* Program the randomisation service
* Develop a database
Apr 01--------------------------------------Dec 04
* Recruitment in all centres
Apr 01-------------------------------------------------------Sep 07
* Data collection and management
Oct 02-----------------------May 07
* 18-24 month follow-up
Sep--Dec 05
* Final analysis of primary outcomes
Sep--Dec 07
* Final analysis of 18-24 month follow-up and secondary analyses
2.15 Committee Structure
The
Steering Committee meets every 2-3 months. The committee is responsible for decisions related to the organisation and conduct of the trial. The steering committee consists of the following members:
Dr Kellie Murphy is the Principal Investigator and is responsible for the distribution of funds, and for the overall progress and timely completion of the trial. She is also responsible for the liaison with the clinical centres and for responding to clinical questions relating to the trial protocol. Dr Mary Hannah is responsible for supervising the activities of the Data Centre personnel and for assisting with the liaison with the clinical centres. Dr Susan Ross is responsible for providing advice on regulatory issues. Ms Sheila Hewson is responsible for providing advice on trial administration and data management. The trial coordinator is responsible for overseeing the data management, maintaining contact with the various collaborating centres and the day to day trial activities. Dr Arne Ohlsson, Dr Saroj Saigal, Dr Ed Kelly, and Dr Shoo Lee are responsible for providing advice on the neonatal, infant and neurological (18-24 month) outcomes. They are also be responsible for assisting with the liaison with the clinical centres. Dr Kofi Amankwah, Dr Marie-France Delisle, and Dr Cindy Maxwell are responsible for providing obstetrical input and for assisting with the liaison with the clinical centres. Dr Stephen Matthews is responsible for providing advice on the neurological (18-24) outcomes and also for input from a basic scientist’s perspective. Ms Patricia Guselle is a consumer who provides input on all aspects of the conduct of the study. Dr`Elizabeth Asztalos, Dr Joanne Rovet, Dr Marcia Barnes, and Dr Renee Sananes are responsible for planning the 5 year follow-up assessments. Dr Andrew Willan is responsible for the statistical analyses and Dr Amiram Gafni is responsible for the economic analysis. Dr Fariba Aghajafari was responsible for assisting with the development of the data forms and with the manual of operations for the centres at the beginning of the study.
The
Collaborative Group consists of the Steering Committee, the obstetrical investigators and the clinical coordinators at the centres. This group provides input on all major issues regarding the trial.
The
Data and Safety Monitoring Board consists of Dr Michael Bracken, Professor, Department of Epidemiology and Public Health, Yale University School of Medicine (chair); Dr Jon Tyson, Professor, Department of Pediatrics, University of Texas Health Science Center at Dallas; and Dr Lelia Duley, Resource Centre for Randomised Trials, Nuffield Department of Clinical Medicine, University of Oxford. This committee will review the results of the interim analysis as well as any other data the Steering Committee is concerned about and will recommend early termination of the trial if appropriate.
2.16 Data Management and Validation
Women will be assessed to confirm eligibility and baseline information will be collected and transmitted to the Data Centre during the randomisation telephone call. Outcome and other descriptive data will be collected on trial data forms and will be mailed to the Data Centre. Data will be scanned into a “Teleform” data management system. Logic and range checks will verify the accuracy of the data. Data forms with errors or missing data will be returned to the clinical centres for correction and/or completion. Data will be managed according to Good Clinical Practice Guidelines.43
2.17 Relevance
In Canada today, approximately 7% of infants are born preterm.2 It is well known that these infants account for a disproportionate amount of neonatal morbidity and mortality.1 Some of these infants are exposed to multiple courses of ACS despite the fact that the benefit-to-risk ratio has not been established. In this trial, if multiple courses of ACS are found to be beneficial it will lead to a decrease in neonatal morbidity and mortality and will be cost saving. If there is no difference between single vs. multiple courses of ACS, then this treatment should not be given and this will be cost saving. Finally, if multiple courses of ACS are found to be detrimental, then practice patterns will have to change and this will benefit infants and children and will ultimately lead to a cost saving as well. An appropriately-sized and well-designed RCT is needed to determine the effectiveness of this important antenatal treatment.
3. REFERENCES
- Berkowitz GS, Papiernik E. Epidemiology of preterm birth. Epidemiology Review 1993;15:414-43.
- Joseph KS, Kramer MS, Marcoux S, Ohlsson A, Wen SW, Allen A, Platt R. Determinants of preterm birth rates in Canada from 1981 through 1983 and from 1992 through 1994. N Engl J Med 1998;339:1434-9.
- Liggins GC, Howie RN. A controlled trial of antepartum glucocorticoid treatment for prevention of the respiratory distress syndrome in premature infants. Pediatrics 1972;50:515-25.
- Crowley P, Chalmers I, Keirse MJNC. The effects of corticosteroid administration before preterm delivery: an overview of the evidence from controlled trials. Br J Obstet Gynaecol 1990;97:11-25.
- National Institute of Child Health and Human Development Office of Medical Applications of Research, NIH. Report of the Consensus Development Conference on the effect of corticosteroids for fetal maturation on perinatal outcomes. Vol.95-3784: NIH Publication, November, 1994. http://www.nichd.nih.gov/publications/pubs/corticosteroids/Corticosteroids.html
- Ballard PL. Hormonal regulation of pulmonary surfactant. Endocr Rev 1989;10:165-81.
- Lanteri CJ, Willet KE, Kano S, Jobe AH, Ikegami M, Polk DH, Newnham JP, Kohan R, Kelly R, Sly PD. Time course of changes in lung mechanics following fetal steroid treatment. Am J Respir Crit Care Med 1994;150:759-65.
- Ballard PL, Ballard RA. Scientific basis and therapeutic regimens for use of antenatal glucocorticoids. Am J Obstet Gynecol 1995;173:254-64.
- Taeusch HW, Brown E, Torday JS, Nielsen HC. Magnitude and duration of lung response to dexamethasone in fetal sheep Am J Obstet Gynecol 1981;140:452-5.
- Ballard PL, Ertsey R, Gonzales LW, Gonzales J. Transcriptional regulation of human pulmonary surfactant proteins SP-B and SP-C by glucocorticoids. Am J Respir Cell Mol Biol 1996;14:599-607.
- Hanes RC. The pharmacological basis of therapeutics. In: Goodman LS, Gilman A, Rall TW, et al, eds. New York: Pergamon, 1990:1431-59.
- Ballard PL, Granberg JP, Ballard RA. Glucocorticoid levels in maternal and cord serum after prenatal betamethasone therapy to prevent respiratory distress syndrome. J Clin Invest 1975;56:1548-54.
- Crowley P. Corticosteroids prior to preterm delivery. Cochrane Database of Systematic Reviews 2003;3.
- MacArthur BA, Howie RN, Dezoete JA, Elkins J. Cognitive and psychosocial development of 4-year-old children whose mothers were treated antenatally with betamethasone. Pediatr 1981;68:638-43.
- MacArthur BA, Howie RN, Dezoete JA, Elkins J. School progress and cognitive development of 6-year-old children whose mothers were treated antenatally with betamethasone. Pediatr 1982;70:99-105.
- Schmand B, Neuvel J, Smolders-de Haas H, Hoeks J, Treffers PE, Koppe JG. Psychological development of children who were treated antenatally with corticosteroids to prevent respiratory distress syndrome. Pediatr 1990;86:58-64.
- Smolders-de Haas H, Neuvel J, Schmand B, Treffers PE, Koppe JG, Hoeks J. Physical development and medical history of children who were treated antenatally with corticosteroids to prevent respiratory distress syndrome: a 10 to 12 year follow-up. Pediatr 1990;86:65-70.
- Dessens AB, Smolders-de Haas, Koppe JG. Twenty-year follow-up of antenatal corticosteroid treatment. Pediatrics 2000;105(6). URL: http://www.pediatrics.org/cgi/content/full/105/6/e77.
- Collaborative Group on Antenatal Steroids Therapy: Effects of antenatal dexamethasone administration in the infant: Long-term follow-up. J Pediatr 1984;104:259-67.
- Taeusch HW, Frigoletto F, Kitzmiller J, Avery ME, Hehre A, Fromm B, Lawson E, Neff RK. Risk of respiratory distress syndrome after prenatal dexamethasone treatment. Pediatr 1979;63:64-72.
- Collaborative Group on Antenatal Steroid Therapy. Effect of antenatal dexamethasone administration on the prevention of respiratory distress syndrome. Am J Obstet Gynecol 1981;141:276-86.
- Wallace EM, Chapman J, Stenson B, Wright S. Antenatal corticosteroid prescribing: setting standards of care. Br J Obstet Gynaecol 1997;104:1262-6.
- Brocklehurst P, Gates S, McKenzie-McHarg K, Alfirevic Z, Chamberlain G. Are we prescribing multiple courses of antenatal corticosteroids? A survey of practice in the UK. Br J Obstet Gynaecol 1999;106:977-9.
- Quinlivan JA, Evans SF, Dunlop SA, Beazley LD, Newnham JP. Use of corticosteroids by Australian obstetricians - a survey of clinical practice. Aust NZ J Obstet Gynaecol 1998;38:1-7.
- Guinn DA, Atkinson MW, Sullivan L et al. Single vs weekly courses of antenatal corticosteroids for women at risk of preterm delivery: A randomized controlled trial. JAMA 2001;286(13):1581-7.
- Aghajafari F, Murphy K, Ohlsson A, Amankwah K, Matthews S, Hannah M. Multiple versus single courses of antenatal corticosteroids for preterm birth: a pilot study. JOGC 2002;24:321-9.
- McEvoy C, Bowling S, Williamson K, Lozano D, Tolaymat L, Izquierdo L, Maher J, Helfgott A. The Effect of a single remote course versus weekly courses of antenatal corticosteroids on functional residual capacity in preterm infants: a randomized trial. Pediatr 2002;110:280-4.
- Crowther CA, Harding J. Repeat doses of prenatal corticosteroids for women at risk of preterm birth for preventing neonatal respiratory disease. Cochrane Database of Systematic Reviews 2003;3.
- Aghajafari F, Murphy K, Willan A, Ohlsson A, Amankwah K, Matthews S, Hannah M. Multiple courses of antenatal corticosteroids: a systematic review and meta-analysis. Am J Obstet Gynecol 2001;185:1073-80.
- Thorp JA, O’Connor M, Belden B, Etzenhouser J, Hoffman EL, Jones PG. Effects of phenobarbital and multiple-dose corticosteroids on developmental outcome at age 7 years. Obstet Gynecol 2003;101:363-73.
- Aghajafari F, Murphy K, Matthews S, Ohlsson A, Amankwah K, Hannah M. Repeated doses of antenatal corticosteroids in animals: a systematic review. Am J Obstet Gynecol 2002;186:843-9.
- Levitt NS, Lindsay RS, Holmes MC, Seckl JR. Dexamethasone in the last week of pregnancy attenuates hippocampal glucocorticoid receptor gene expression and elevates blood pressure in the adult offspring in the rat. Neuroendocrinology 1996;64:412-8.
- Dean F, Matthews SG. Maternal dexamethasone treatment in late gestation alters glucocorticoid and mineralocorticoid receptor mRNA in the fetal guinea pig brain. Brain Research 1999;846:253-9.
- Halliday HL, Ehrenkranz RA, Doyle LW. Early postnatal (< 96 hours) corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews 2003;3.
- Hennekens CH, Buring JE. Epidemiology in medicine. Boston: Little Brown and Company, 1987:178.
- Palisano R, Rosenbaum P, Walter S, Russell D, Wood E, Galuppi B. Development and reliability of a system to classify gross motor function in children with cerebral palsy. Dev Med Child Neurol 1997;39(4):214-23.
- Bayley N. Bayley Scales of Infant Development. 2nd Edition San Antonio, TX: Psychological Corporation;1993.
- Papile LA, Burstein J, Burstein R, Koffler H. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weight less than 1500 grams. J Pediatr 1978;92:529-34.
- Walsh MC, Kliegman RM. Necrotizing enterocolitis: Treatment based on staging criteria. Pediatr Clin of North Am 1986;179:33.
- Cox JL, Holden JM, Detection of postnatal depression, development of the 10-item Edinburgh postnatal depression scale. Br J Psych 1987;150:782-6.
- Ballard RA, Ballard PL, Cnaan A, Pinto-Martin J, David DJ, Padbury JF, Phibbs R, Parer JT, Hart MC, Mannino FL, Sawai SK. Antenatal thyrotropin-releasing hormone to prevent lung disease in preterm infants. N Engl J Med 1998;338:493-8.
- Knight DB, Liggins GC, Wealthall SR. A randomised, controlled trial of antepartum thyrotropin-releasing hormone and betamethasone in the prevention of respiratory disease in preterm infants. Am J Obstet Gynecol 1994;171:11-6.
- Good Clinical Practice: Consolidated Guideline. Minister of Health, Ottawa, 1997.


