| 1. | Genetic Networks: Large-Scale Mapping of Synthetic Lethal Interactions | |
| a. | Mapping Genetic Interaction Networks | |
| b. | Genetic Interaction Data - Imaging, Processing, and Analysis | |
| c. | Temperature Sensitive Conditional Mutant Collection | |
| d. | Modeling Genetic Networks | |
| 2. | Sigma Deletion Mutant Collection | |
| 3. | High-Content Cell Biology | |
| 4. | Chemical-Genetics | |
| a. | Comparing Chemical-Genetic & Genetic Interaction Networks | |
| b. | Barcoded Yeast Gene Library: Drug-Resistant Mutants and Dosage Suppression | |
| 5. | Mapping Protein-Protein Interaction Networks for Peptide Recognition Modules | |
Genetic Networks:
Mapping Genetic Interaction Networks
Collaborative Project with Brenda Andrews' Lab
People
Michael Costanzo, Taras Makhnevych, Hong Xu, Renee Brost, Yiqun Chen, Hongwei Zhu, Jadine Paw, Michaela Marback, Hong Lu, Bryan-Joseph San Luis, Nadia Remtulla, Charly Chahwan
The genetic makeup of an individual is extremely complex owing to the collaborative interaction of particular genes and alleles. When considering inherited diseases, a major challenge is identifying the combination of natural genetic variants that modulate the activity of specific disease-associated pathways. One way to address this challenge is to map allele combinations or genetic networks that control specific phenotypes.
Our laboratory employs the budding yeast, S. cerevisiae, as a model to investigate the basic principles of genetic interaction networks. Most yeast genes are not required for viability indicating that essential processes are regulated by multiple compensatory pathways. Synthetic lethality defines a relationship where two mutations, neither of which is lethal on its own, result in cell death when combined. Identification of synthetically lethal double mutant combinations is of particular interest because they can identify genes whose products impinge on the same essential biological process. An alternate interpretation of synthetic lethality defines a relationship where the presence of one gene allows an organism to tolerate genetic variation in a different gene. Thus, genetic interaction maps provide valuable insights into the concept of genetic robustness, that is, how an organism is able to buffer or tolerate the phenotypic consequences of genetic variation.
Our goal is to map the first complete eukaryotic genetic network. To this end, our laboratory has established methodology and infrastructure to generate a global genetic interaction map. Synthetic genetic array (SGA) analysis is an automated approach enabling the systematic isolation of yeast double mutants and subsequent large-scale mapping of genetic interactions [Tong et al., Science 294:2364-2368 (2001)] (Fig.1 & Fig.2).
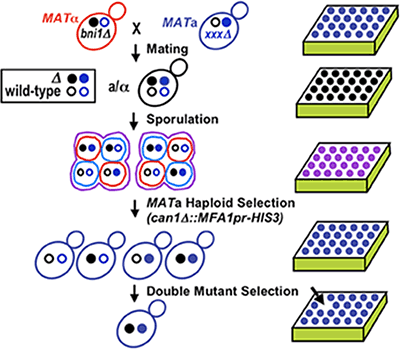
Figure 1
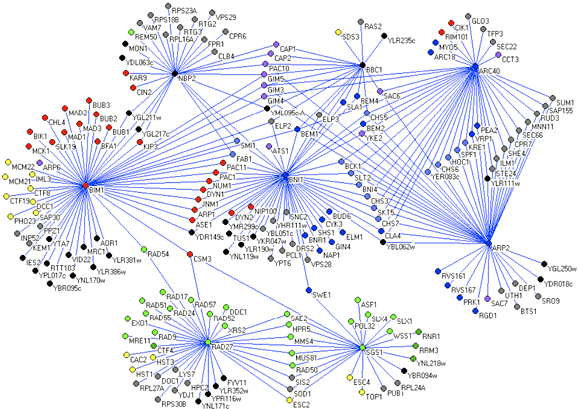
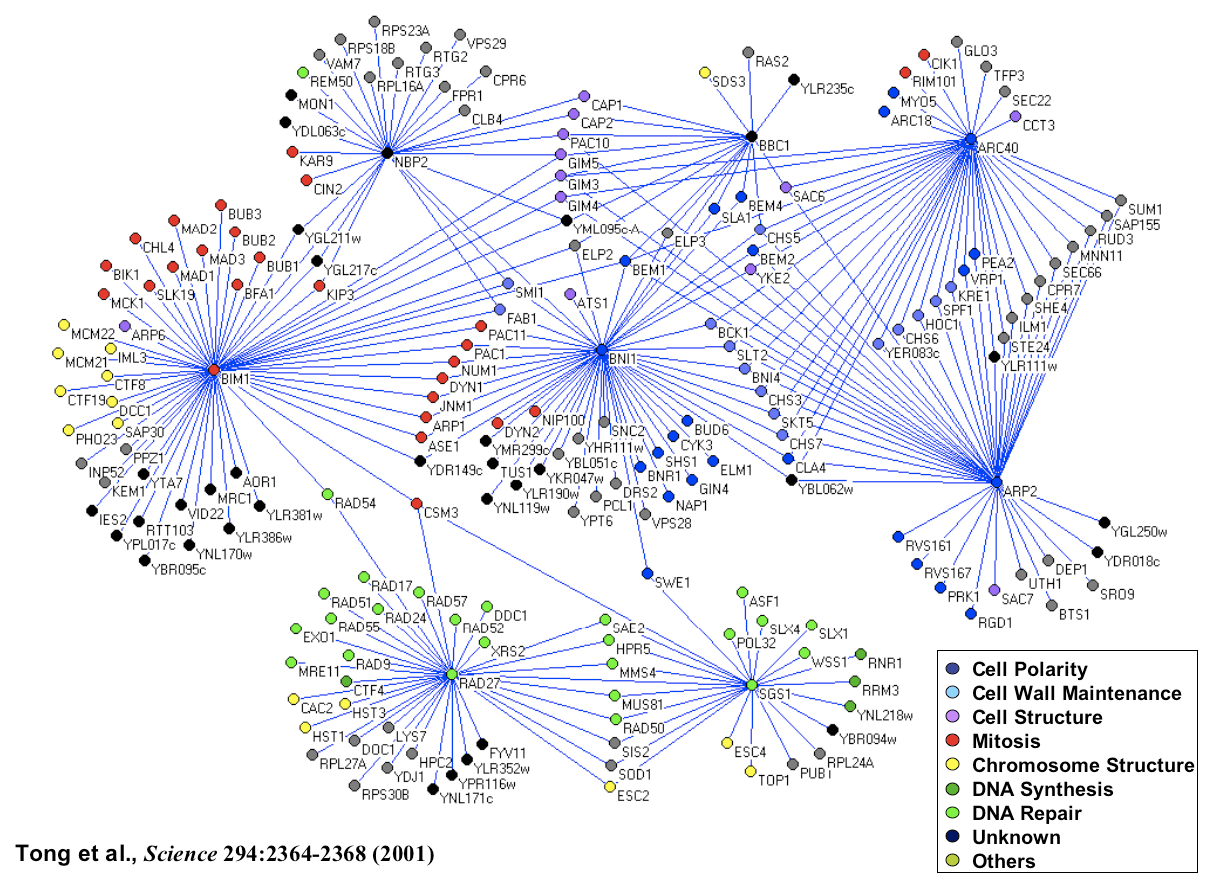
Analysis of our initial network consisting of 1,000 genes and 4,000 interactions revealed that the yeast genetic interaction network, similar to most biological networks, exhibits scale-free, small-world properties. Consistent with such characteristics, genetic interactions do not occur randomly but rather are highly organized whereby genes with similar roles tend to interact with one another in a manner reflecting cellular function. Furthermore, despite being sparse in nature, network position and gene connectivity is highly predictive of novel genetic interactions and gene function [Tong et al., Science 303: 808-813 (2004)]. In particular, genes within the same pathway show similar genetic interaction patterns and, therefore, clustering of genetic interactions sorts gene sets into functional pathways and complexes (Fig.3).
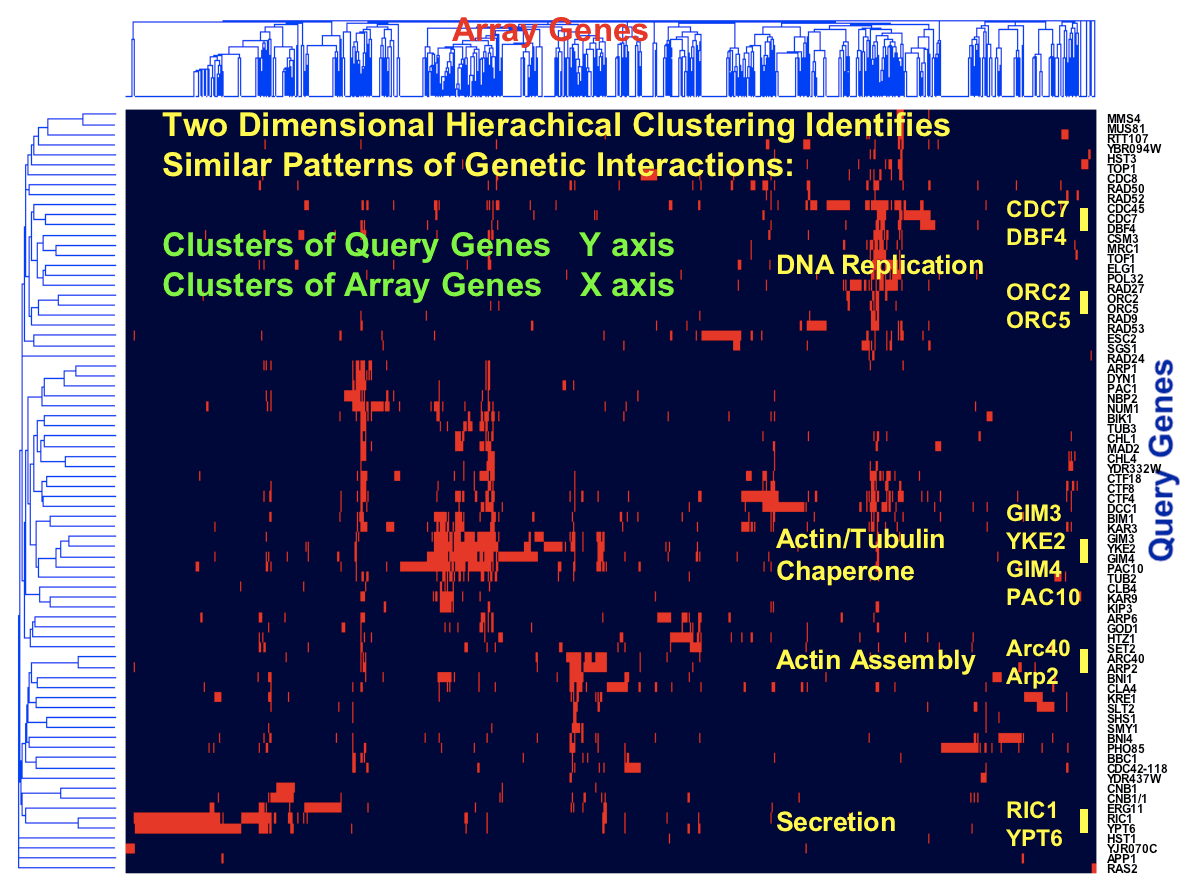
By applying SGA to systematically assess the fitness of all pair-wise double mutant combinations (~18 x 106) in S. cerevisiae, we hope to assign function to uncharacterized genes, identify genetic relationships between biological processes and begin to understand the genetic underpinnings of complex inheritable disease.